
 Yuanlong LIU
912b2b6f3b
Update README.md
Yuanlong LIU
912b2b6f3b
Update README.md
|
4 vuotta sitten | |
|---|---|---|
| R | 4 vuotta sitten | |
| img | 4 vuotta sitten | |
| inst | 4 vuotta sitten | |
| man | 4 vuotta sitten | |
| src | 4 vuotta sitten | |
| .DS_Store | 4 vuotta sitten | |
| .gitattributes | 4 vuotta sitten | |
| .gitignore | 4 vuotta sitten | |
| DESCRIPTION | 4 vuotta sitten | |
| LICENSE | 4 vuotta sitten | |
| NAMESPACE | 4 vuotta sitten | |
| README.md | 4 vuotta sitten |
README.md
CALDER user manuel
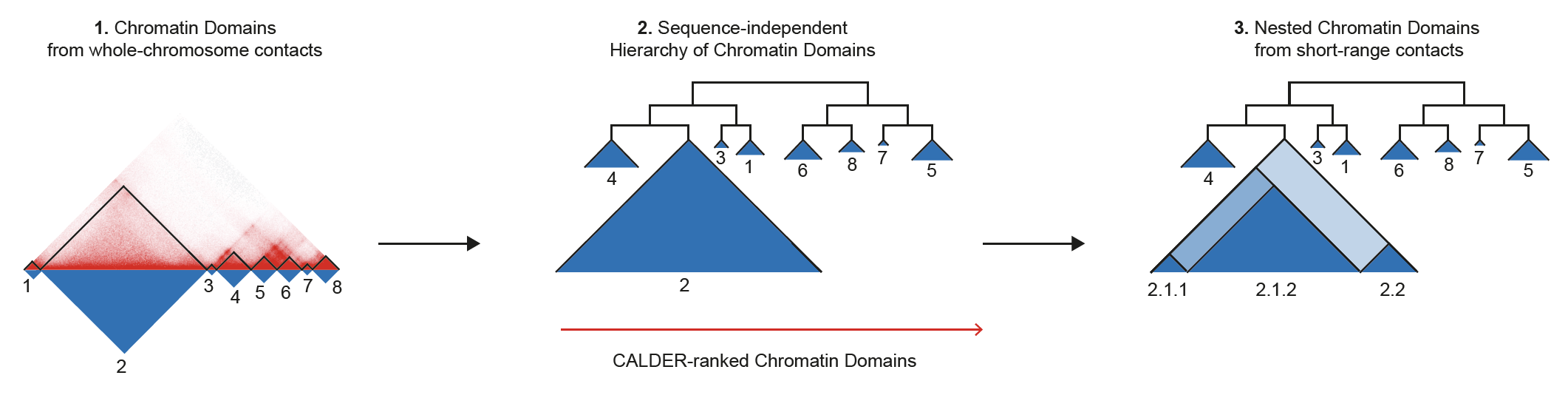
CALDER is a Hi-C analysis tool that allows: (1) compute chromatin domains from whole chromosome contacts; (2) derive their non-linear hierarchical organization and obtain sub-compartments; (3) compute nested sub-domains within each chromatin domain from short-range contacts. CALDER is currently implemented in R.
We added multiple new features in version 2.0
- Support for hg19, hg38, mm9, mm10 and other genomes
- Support input in .hic format generated by Juicer tools (https://github.com/aidenlab/juicer)
- Opimized resolution selection
- Added output in tabular .txt format for downstream analysis
- Aggregated all chromosome output into a single file
Installation
Make sure all dependencies have been installed:
- R.utils (>= 2.9.0),
- doParallel (>= 1.0.15),
- ape (>= 5.3),
- dendextend (>= 1.12.0),
- fitdistrplus (>= 1.0.14),
- igraph (>= 1.2.4.1),
- Matrix (>= 1.2.17),
- rARPACK (>= 0.11.0),
- factoextra (>= 1.0.5),
- maptools (>= 0.9.5),
- data.table (>= 1.12.2),
- fields (>= 9.8.3),
- GenomicRanges (>= 1.36.0)
- ggplot2 (>= 3.3.5)
- strawr (>= 0.0.9)
Clone its repository and install it from source:
git clone https://github.com/CSOgroup/CALDER.git
install.packages(path_to_CALDER, repos = NULL, type="source") ## install from the cloned source file
Please contact yliueagle@googlemail.com for any questions about installation.
install CALDER and dependencies automaticly:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("GenomicRanges")
install.packages("remotes")
remotes::install_github("CSOgroup/CALDER")
Usage
The input data of CALDER is a three-column text file storing the contact table of a full chromosome (zipped format is acceptable, as long as it can be read by data.table::fread). Each row represents a contact record pos_x, pos_y, contact_value, which is the same format as that generated by the dump command of juicer (https://github.com/aidenlab/juicer/wiki/Data-Extraction):
16050000 16050000 10106.306
16050000 16060000 2259.247
16060000 16060000 7748.551
16050000 16070000 1251.3663
16060000 16070000 4456.1245
16070000 16070000 4211.7393
16050000 16080000 522.0705
16060000 16080000 983.1761
16070000 16080000 1996.749
...
A demo dataset is included in the repository CALDER/inst/extdata/mat_chr22_10kb_ob.txt.gz and can be accessed by system.file("extdata", "mat_chr22_10kb_ob.txt.gz", package='CALDER') once CALDER is installed. This data contains contact values of GM12878 on chr22 binned at 10kb (Rao et al. 2014)
CALDER contains three modules: (1) compute chromatin domains; (2) derive their hierarchical organization and obtain sub-compartments; (3) compute nested sub-domains within each compartment domain.
To run three modules in a single step:
CALDER_main(contact_mat_file,
chr,
bin_size,
out_dir,
sub_domains=TRUE,
save_intermediate_data=FALSE,
genome='hg19')
To run three modules in seperated steps:
# This will not compute sub-domains, but save the intermediate_data that can be used to compute sub-domains latter on
CALDER_main(contact_mat_file,
chr,
bin_size,
out_dir,
sub_domains=FALSE,
save_intermediate_data=TRUE,
genome='hg19')
# (optional depends on needs) Compute sub-domains using intermediate_data_file that was previous saved in the out_dir (named as chrxx_intermediate_data.Rds)
CALDER_sub_domains(intermediate_data_file,
chr,
out_dir,
bin_size)
Paramters:
contact_mat_file: path to the contact table of a chromosomechr: chromosome number. Either numeric or character, will be pasted to the output namebin_size: numeric, the size of a bin in consistent with the contact tableout_dir: the output directorysub_domains: logical, whether to compute nested sub-domainssave_intermediate_data: logical. If TRUE, an intermediate_data will be saved. This file can be used for computing nested sub-domains later ongenome: string. Specifies the genome assembly (Default="hg19").
| contact_tab_straw | |
| ref_genome | One of 'hg19', 'hg38', 'mm9', 'mm10', or 'others' by default. |
| annotation_track | A table in data.frame or data.table format |
| contact_file_hic | Path to the hic file |
| subcomp | Optional: Assigning sub-compartments based on PC magnitude values using HMM segmentation |
| fithic | Run Fit-Hi-C to identify loops before running dloop (Optional but recommended) |
| dloop | Find differential loops anchored in at least one of the differential compartments across the samples (Optional but recommended) |
| viz | Generate IGV vizualization HTML file. Must have performed other steps in order (optional ones not strictly necessary) before this one. |
| enrich | Perform gene enrichment analysis (GSEA) of genes in differential compartments/loops |
Output:
chrxx_domain_hierachy.tsv
- information of compartment domain and their hierarchical organization. The hierarchical structure is fully represented by
compartment_label, for example,B.2.2.2andB.2.2.1are two sub-branches ofB.2.2. Thepos_endcolumn specifies all compartment domain borders, except when it is marked asgap, which indicates it is the border of a gap chromsome region that has too few contacts and was excluded from the analysis (e.g., due to low mappability, deletion, technique flaw)
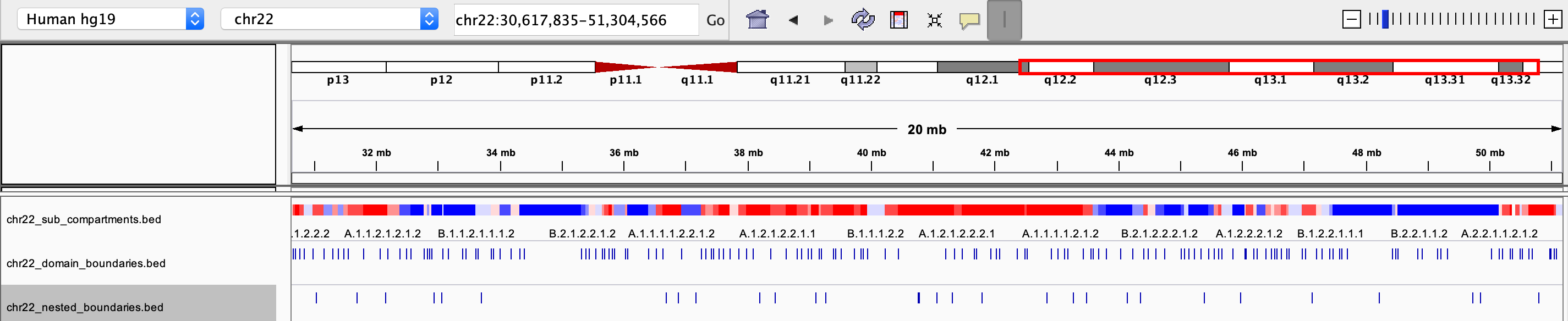
chrxx_sub_compartments.bed
- a .bed file containing the sub-compartment information, that can be visualized in IGV. Different colors were used to distinguish compartments (at the resolution of 8 sub-compartments)
chrxx_domain_boundaries.bed
- a .bed file containing the chromatin domains boundaries, that can be visualized in IGV
chrxx_nested_boundaries.bed
- a .bed file containing the nested sub-domain boundaries, that can be visualized in IGV
chrxx_intermediate_data.Rds
- an Rds file storing the intermediate_data that can be used to compute nested sub-domains (if CALDER is run in two seperated steps)
chrxx_log.txt, chrxx_sub_domains_log.txt
- log file storing the status and running time of each step
Runnig time:
For the computational requirement, running CALDER on the GM12878 Hi-C dataset at bin size of 40kb took 36 minutes to derive the chromatin domains and their hierarchy for all chromosomes (i.e., CALDER Step1 and Step2); 13 minutes to derive the nested sub-domains (i.e., CALDER Step3). At the bin size of 10kb, it took 1 h 44 minutes and 55 minutes correspondingly (server information: 40 cores, 64GB Ram, Intel(R) Xeon(R) Silver 4210 CPU @ 2.20GHz). The evaluation was done using a single core although CALDER can be run in a parallel manner.
Demo run:
library(CALDER)
contact_mat_file = system.file("extdata", "mat_chr22_10kb_ob.txt.gz", package = 'CALDER')
CALDER_main(contact_mat_file, chr=22, bin_size=10E3, out_dir='./GM12878', sub_domains=TRUE, save_intermediate_data=FALSE)
The saved .bed files can be view directly through IGV:
Citation
If you use CALDER in your work, please cite: https://www.nature.com/articles/s41467-021-22666-3
Contact information
- Author: Yuanlong LIU
- Affiliation: Computational Systems Oncology group, Department of Computational Biology, University of Lausanne, Switzerland
- Email: yliueagle@googlemail.com