
README.md 12 KB
CALDER user manuel
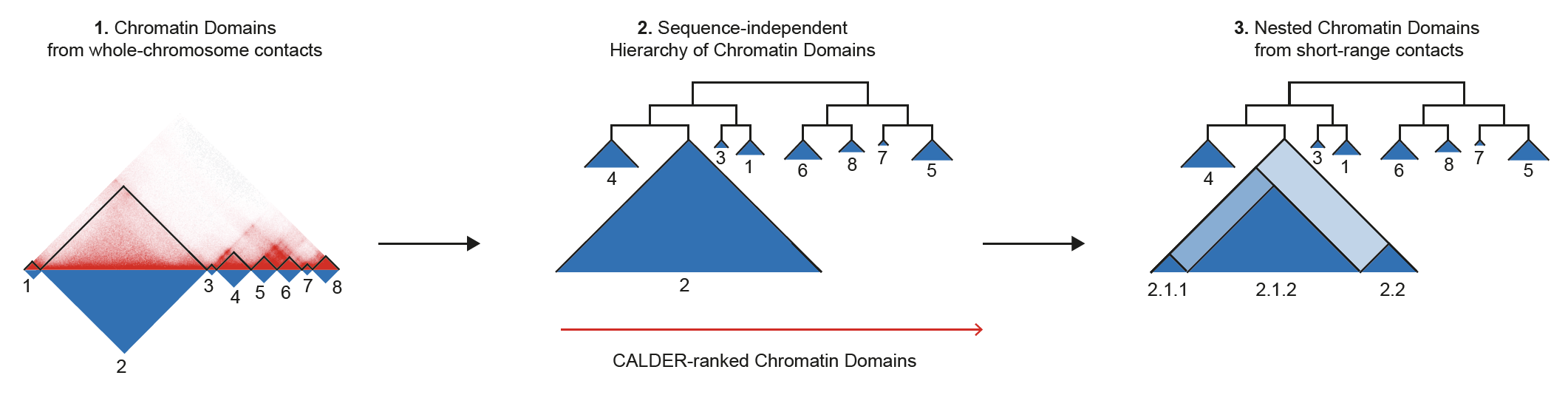
CALDER is a Hi-C analysis tool that allows: (1) compute chromatin domains from whole chromosome contacts; (2) derive their non-linear hierarchical organization and obtain sub-compartments; (3) compute nested sub-domains within each chromatin domain from short-range contacts. CALDER is currently implemented in R.
Multiple new features were added in version 2.0
- Support for hg19, hg38, mm9, mm10 and other genomes
- Support input in .hic format generated by Juicer tools (https://github.com/aidenlab/juicer)
- Opitimized bin_size selection for more reliable compartment identification
- Aggregated all chromosome output into a single file for easier visualization in IGV
- Added output in tabular .txt format at bin level for easier downstream analysis
Introduction of opitimized bin_size selection
Due to reasons such as low data quality or large scale structrual variation, compartments can be unreliablly called at one bin_size (equivalent to resoltution in the literature) but correctly called at another bin_size. We added an opitimized bin_size selection strategy to call reliable compartments. This strategey is based on the observation from our large scale compartment analysis (https://www.nature.com/articles/s41467-021-22666-3) that, although compartments can change between different conditions, their overall correlation cor(compartment_rank_1, compartment_rank_2) is high (> 0.4).
The strategy: given a bin_size specified by user, we call compartments with extended bin_sizes and choose the smallest bin_size such that no bigger bin_size can increase the compartment correclation with a reference compartment more than 0.05. For example, if correclation for bin_size=10000 is 0.2 while for bin_size=50000 is 0.6, we are more confident the latter is more reliable; if correclation for bin_size=10000 is 0.5 while for bin_size=50000 is 0.52, we would choose the former as it has higher resolution.
bin_size is extended in the following way thus to aggregate directly from the input contact matrix into larger bin_sizes, without the need to provide additional contact matrices from user side
if(bin_size==5E3) bin_sizes = c(5E3, 10E3, 50E3, 100E3)
if(bin_size==10E3) bin_sizes = c(10E3, 50E3, 100E3)
if(bin_size==20E3) bin_sizes = c(20E3, 40E3, 100E3)
if(bin_size==25E3) bin_sizes = c(25E3, 50E3, 100E3)
if(bin_size==40E3) bin_sizes = c(40E3, 80E3)
if(bin_size==50E3) bin_sizes = c(50E3, 100E3)
Note that this strategy is currently only available for hg19, hg38, mm9 and mm10 genome for which we generated high quality reference compartments using Hi-C data from: GSE63525 for hg19, https://data.4dnucleome.org/files-processed/4DNFI1UEG1HD/ for hg38, GSM3959427 for mm9, http://hicfiles.s3.amazonaws.com/external/bonev/CN_mapq30.hic for mm10.
Introduction of Calder analysis for other genomes
Although CALDER was mainly tested on human and mouse dataset, it can be applied on dataset from other genomes. One additional information is required in such case: a feature_track that is presumably positively correlated with compartment score (thus higher values in A than in B compartment). This information will be used for correctly determing the A/B direction. Some suggested tracks are gene density, H3K27ac, H3K4me1, H3K4me2, H3K4me3, H3K36me3 (or negative transform of H3K9me3) signals. Note that this information will not alter the hierarchical compartment/TAD structure, and can come from any external study with matched genome.
Installation
Make sure all dependencies have been installed:
- R.utils (>= 2.9.0),
- doParallel (>= 1.0.15),
- ape (>= 5.3),
- dendextend (>= 1.12.0),
- fitdistrplus (>= 1.0.14),
- igraph (>= 1.2.4.1),
- Matrix (>= 1.2.17),
- rARPACK (>= 0.11.0),
- factoextra (>= 1.0.5),
- maptools (>= 0.9.5),
- data.table (>= 1.12.2),
- fields (>= 9.8.3),
- GenomicRanges (>= 1.36.0)
- ggplot2 (>= 3.3.5)
- strawr (>= 0.0.9)
Clone its repository and install it from source:
git clone https://github.com/CSOgroup/CALDER.git
install.packages(path_to_CALDER, repos = NULL, type="source") ## install from the cloned source file
Please contact yliueagle@googlemail.com for any questions about installation.
install CALDER and dependencies automaticly:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("GenomicRanges")
install.packages("remotes")
remotes::install_github("CSOgroup/CALDER")
Usage
The input data of CALDER is a three-column text file storing the contact table of a full chromosome (zipped format is acceptable, as long as it can be read by data.table::fread). Each row represents a contact record pos_x, pos_y, contact_value, which is the same format as that generated by the dump command of juicer (https://github.com/aidenlab/juicer/wiki/Data-Extraction):
16050000 16050000 10106.306
16050000 16060000 2259.247
16060000 16060000 7748.551
16050000 16070000 1251.3663
16060000 16070000 4456.1245
16070000 16070000 4211.7393
16050000 16080000 522.0705
16060000 16080000 983.1761
16070000 16080000 1996.749
...
A demo dataset is included in the repository CALDER/inst/extdata/mat_chr22_10kb_ob.txt.gz and can be accessed by system.file("extdata", "mat_chr22_10kb_ob.txt.gz", package='CALDER') once CALDER is installed. This data contains contact values of GM12878 on chr22 binned at 10kb (Rao et al. 2014)
CALDER contains three modules: (1) compute chromatin domains; (2) derive their hierarchical organization and obtain sub-compartments; (3) compute nested sub-domains within each compartment domain.
To run three modules in a single step:
CALDER_main(contact_mat_file,
chr,
bin_size,
out_dir,
sub_domains=TRUE,
save_intermediate_data=FALSE,
genome='hg19')
To run three modules in seperated steps:
# This will not compute sub-domains, but save the intermediate_data that can be used to compute sub-domains latter on
CALDER_main(contact_mat_file,
chr,
bin_size,
out_dir,
sub_domains=FALSE,
save_intermediate_data=TRUE,
genome='hg19')
# (optional depends on needs) Compute sub-domains using intermediate_data_file that was previous saved in the out_dir (named as chrxx_intermediate_data.Rds)
CALDER_sub_domains(intermediate_data_file,
chr,
out_dir,
bin_size)
Paramters:
| Name | Description |
| --------------------- | ----------------------- |
| chrs | A vector of chromosome names to be analyzed, with or without 'chr'
| contact_file_dump |A list of contact files in dump format, named by chrs. Each contact file stores the contact information of the corresponding chr. Only one of contact_file_dump, contact_tab_dump, contact_file_hic should be provided
| contact_tab_dump | A list of contact table in dump format, named by chrs, stored as an R object. Only one of contact_file_dump, contact_tab_dump, contact_file_hic should be provided
| contact_file_hic | A hic file generated by Juicer tools. It should contain all chromosomes in chrs. Only one of contact_file_dump, contact_tab_dump, contact_file_hic should be provided
| ref_genome | One of 'hg19', 'hg38', 'mm9', 'mm10', 'others' (default). These compartments will be used as reference compartments for optimized bin_size selection. If ref_genome = others, a feature_track should be provided (see below) and no optimized bin_size selection will be performed
| feature_track | A genomic annotation track in data.frame or data.table format. This track will be used for determing the A/B compartment direction when ref_genome=others and should presumably have higher values in A than in B compartment. Some suggested tracks can be gene density, H3K27ac, H3K4me1, H3K4me2, H3K4me3, H3K36me3 (or negative transform of H3K9me3 signals)
| bin_size | The bin_size (resolution) to run CALDER. bin_size should be consistent with the data resolution in contact_file_dump or contact_tab_dump if these files are provided as input, otherwise bin_size should exist in the contact_file_hic file. Recommended bin_size is between 10000 to 50000
| save_dir | the directory to save outputs
| save_intermediate_data | logical. If TRUE, an intermediate_data will be saved. This file can be used for computing nested sub-domains later on
| n_cores | integer. Number of cores to be registered for running CALDER in parallel
| single_binsize_only | logical. If TRUE, CALDER will compute compartments only using the bin_size specified by the user and not do bin size optimization
| sub_domains | logical, whether to compute nested sub-domains
Output:
chrxx_domain_hierachy.tsv
- information of compartment domain and their hierarchical organization. The hierarchical structure is fully represented by
compartment_label, for example,B.2.2.2andB.2.2.1are two sub-branches ofB.2.2. Thepos_endcolumn specifies all compartment domain borders, except when it is marked asgap, which indicates it is the border of a gap chromsome region that has too few contacts and was excluded from the analysis (e.g., due to low mappability, deletion, technique flaw)
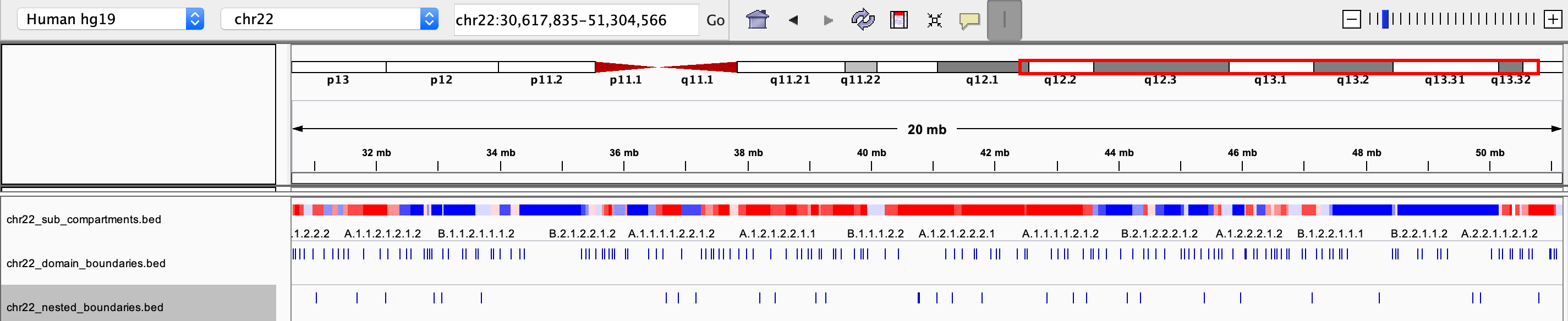
chrxx_sub_compartments.bed
- a .bed file containing the sub-compartment information, that can be visualized in IGV. Different colors were used to distinguish compartments (at the resolution of 8 sub-compartments)
chrxx_domain_boundaries.bed
- a .bed file containing the chromatin domains boundaries, that can be visualized in IGV
chrxx_nested_boundaries.bed
- a .bed file containing the nested sub-domain boundaries, that can be visualized in IGV
chrxx_intermediate_data.Rds
- an Rds file storing the intermediate_data that can be used to compute nested sub-domains (if CALDER is run in two seperated steps)
chrxx_log.txt, chrxx_sub_domains_log.txt
- log file storing the status and running time of each step
Output Structure
The output of the workflow is stored in the folder specified by --save_dir ("results" by default) and will look like this:
results/
└── HiC_sample_1
├── 100000
│ └── KR
│ ├── chr1
│ │ ├── chr1_domain_boundaries.bed
│ │ ├── chr1_domain_hierachy.tsv
│ │ ├── chr1_log.txt
│ │ ├── chr1_nested_boundaries.bed
│ │ ├── chr1_sub_compartments.bed
│ │ └── chr1_sub_domains_log.txt
Runnig time:
For the computational requirement, running CALDER on the GM12878 Hi-C dataset at bin size of 40kb took 36 minutes to derive the chromatin domains and their hierarchy for all chromosomes (i.e., CALDER Step1 and Step2); 13 minutes to derive the nested sub-domains (i.e., CALDER Step3). At the bin size of 10kb, it took 1 h 44 minutes and 55 minutes correspondingly (server information: 40 cores, 64GB Ram, Intel(R) Xeon(R) Silver 4210 CPU @ 2.20GHz). The evaluation was done using a single core although CALDER can be run in a parallel manner.
Demo run:
library(CALDER)
contact_mat_file = system.file("extdata", "mat_chr22_10kb_ob.txt.gz", package = 'CALDER')
CALDER_main(contact_mat_file, chr=22, bin_size=10E3, out_dir='./GM12878', sub_domains=TRUE, save_intermediate_data=FALSE)
The saved .bed files can be view directly through IGV:
Citation
If you use CALDER in your work, please cite: https://www.nature.com/articles/s41467-021-22666-3
Contact information
- Author: Yuanlong LIU
- Affiliation: Computational Systems Oncology group, Department of Computational Biology, University of Lausanne, Switzerland
- Email: yliueagle@googlemail.com